Although regulations often vary by region, regulators intend to protect public health. Regulatory authorities are expected to carry out their public health mandates by facilitating the timely introduction of new medicinal products to meet society’s requirements. At the same time, they are responsible for protecting the public from products of suspect quality, safety, and efficacy.
As outlined above, pharmaceutical products must undergo rigorous regulatory authority evaluation of the dossier or technical file that contains information on quality (chemistry, manufacturing and control (CMC)), safety (nonclinical), and efficacy (clinical) testing results before gaining market access.
With the majority of our clients in the United States, we at Bora work with the regulatory authority known as the US Food and Drug Administration (FDA) who exercise a legal right to control product transport, use, or sale within their jurisdictions. They do this by ensuring compliance with US laws, regulations, standards, and any post-marketing requirements. When a manufacturer (or applicant/ sponsor) plans to market their drugs in the US, a marketing application submission is required. Only after an official application is made to a regulatory authority, can market access be achieved. In general, there are two major types of prescription drug applications to obtain agency product approval, which are NDA and ANDA.
Difference Between NDA and ANDA
 NDA means a New Drug Application. When the sponsor of a new drug believes that enough evidence on the drug’s safety and effectiveness has been obtained to meet the FDA’s requirements for marketing approval, the sponsor submits to the FDA a new drug application (NDA). In other words, when a pharmaceutical company creates a new drug, the company must contact the FDA and demonstrate that the new drug is safe, effective, and meets FDA quality standards. If the NDA is approved, then the product may be marketed in the United States.
NDA means a New Drug Application. When the sponsor of a new drug believes that enough evidence on the drug’s safety and effectiveness has been obtained to meet the FDA’s requirements for marketing approval, the sponsor submits to the FDA a new drug application (NDA). In other words, when a pharmaceutical company creates a new drug, the company must contact the FDA and demonstrate that the new drug is safe, effective, and meets FDA quality standards. If the NDA is approved, then the product may be marketed in the United States.
ANDA means Abbreviated New Drug Application. An abbreviated new drug application (ANDA) contains data that, when submitted to the FDA, provides for the review and ultimate approval of a generic drug product. A generic drug is effectively a copy of an already approved and marketed drug. Generic drug applications are called “abbreviated” because they are not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness. Instead, a generic drug applicant must scientifically demonstrate that its product is bioequivalent (i.e., performs in the same manner as the original drug). Once approved, an applicant may manufacture and market the generic drug product to provide a safe, effective, low-cost alternative versus the branded (original) version to the public.
To this end, an approval letter will be issued when the FDA has determined a drug is safe, effective, can be manufactured acceptably, and be labeled appropriately. An approval is effective on the date the approval letter granting authorization to market the drug for sale in the US is issued.
Changes to an Approved Application
Obtaining approval does not preclude the application holder from making drug product changes. On the contrary, the regulations take into consideration the need for approved application changes. For example, changes in raw material availability, compendial testing updates, technological advances, or manufacturing process improvement needs may require sponsor changes to the approved application.
A drug sponsor should have a robust change management process in place to ensure approved product changes are assessed appropriately for impact and well-documented. A successful change management process will ensure the change’s impact is classified correctly to reduce FDA reclassification risk when filed, which can lead to product distribution delays.
What Should the Concerns for the Change to an Approved FDA NDA and ANDA Be?
The applicant holders of NDAs and ANDAs who intend to make post-approval changes for drugs must assess the effects of the change on the identity, strength (e.g., assay, content uniformity), quality (e.g., physical, chemical, and biological properties), purity (e.g., impurities and degradation products), or potency (e.g., biological activity, bioavailability, bioequivalence) of a drug product as these factors relate to the safety or effectiveness of the drug product. Editorial changes, such as typographical and spelling error corrections or formatting changes to standard operating procedures or batch records, are not required to be submitted.
Fig. 1 summarizes the recommended reporting categories for post-approval changes. Three post-approval change categories are divided: major, moderate, and minor. These changes are categorized based on their potential to affect the drug product’s identity, strength, quality, purity, or potency adversely.
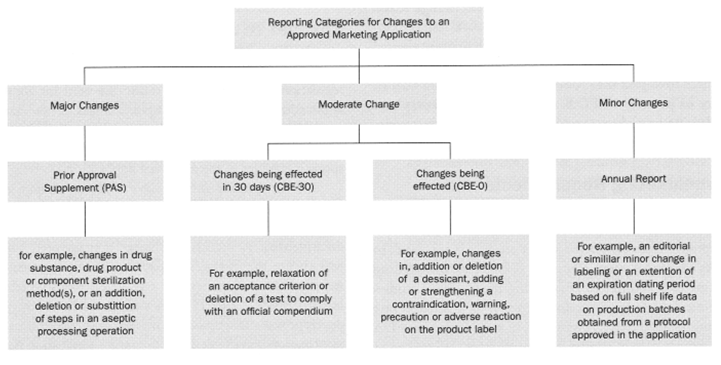
▲Fig.1 (Source: Guidance for Industry: Changes to an Approved NDA or ANDA (April 2004))
1. A Major Change is a change that has a “substantial potential to have an adverse effect” on the identity, strength, quality, purity, or potency of a drug product as these factors may relate to the safety or effectiveness of the drug product. A major change differs from the others in that it requires the submission of a Prior Approval Supplement (PAS), which must be approved by the FDA before distribution of the changed drug product.
2. A Moderate Change is a change that has a “moderate potential to have an adverse effect” on the identity, strength, quality, purity, or potency of the drug product as these factors may relate to the safety or effectiveness of the drug product. There are two types of moderate changes, which include:
-
Change requiring the submission of a Supplement – Changes Being Effected in 30 Days (CBE-30). Changes that fall into this category require the submission of a supplement that must be made to the FDA at least 30 days before the changed drug product is distributed.
-
Change requiring the submission of a Supplement – Changes Being Effected (CBE-0). Changes subject to this type of supplement contain changes for which distribution can occur when FDA receives the supplement.
3. A Minor Change is a change that has “minimal potential to have an adverse effect” on the identity, strength, quality, purity, or potency of the drug product as these factors may relate to the safety or effectiveness of the drug product. There is no submission or supplement required for minor changes, the applicant must simply describe any minor changes that have been made in its next Annual Report (AR) that is submitted to the FDA.
In conclusion, approved marketing application sponsors are required to report almost all changes to FDA under the Federal Food, Drug, and Cosmetic Act (FD&C Act). The Agency has provided numerous guidance documents to aid application holders in determining the appropriate reporting category for post-approval changes to components and composition, manufacturing sites, analytical testing sites, manufacturing processes, specifications, container closure systems, and labeling. These guidance documents also provide advice on the documentation and data to justify changes, as well as handling multiple related changes. The reporting categories are classified based on their potential to impact the product adversely (major- PAS, moderate- CBE-30 or CBE-0, or minor- Annual Report).
However, the application procedure differs from region to region based upon the region’s regulatory body. For international market access, companies must ensure the nonclinical, clinical, CMC, and analytical data generated will satisfy all the regions’ regulatory requirements. Certain specific requirements for tests still may differ, and prospective manufacturers or sponsors should consult with the regulatory authorities in the countries or regions where they intend to market their products.
